
Early anion gap metabolic acidosis in acetaminophen overdose
Original Contribution
Early anion gap metabolic acidosis in acetaminophen overdose
Joe G. Zein MDa, David J. Wallace MD, MPHb,?, Gary Kinasewitz MDc,
Nagib Toubia MDd, Christine Kakoulas MDe
aDivision of Pulmonary and Critical Care, Department of Internal Medicine, Kings County Hospital Center,
Brooklyn, NY 11203, USA
bDepartments of Internal Medicine and Emergency Medicine, Kings County Hospital Center, Brooklyn, NY 11203, USA
cDivision of Pulmonary and Critical Care Medicine, Physiology, and Biophysics, University of Oklahoma Health Science Center,
Box 26901, Oklahoma City, OK 73190, USA
dInternal Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
eYaffe Ruden and Associates, NY 10065, New York, USA
Received 27 January 2009; revised 25 March 2009; accepted 1 April 2009
Abstract
Purpose: The study aimed to determine the incidence and clinical significance of early high (N15 mEq/L) anion gap metabolic acidosis in acetaminophen (APAP) overdose.
Methods: A retrospective review of a cohort of 74 patients presenting within 24 hours of APAP overdose was conducted.
Results: Early high anion gap metabolic acidosis was present in 41% of patients on admission and persisted for 1.5 +- 0.1 days. The anion gap was associated with an elevated lactate level (4.5 +- 1 mmol/L) (r2 = 0.66, P b .05), which persisted for 1 day. The lactate level increased in proportion to the APAP concentration (r2 = 0.75, P b .05). Patients with increased anion gap had a higher incidence of confusion (48% vs 3%; P b.001) and lethargy (39% vs 6%; P = .003). Early high anion gap metabolic acidosis was found in the absence of shock or liver failure. All patients were treated with N-acetylcysteine and, despite the early high anion gap metabolic acidosis, none developed hepatic failure or hypoglycemia.
Conclusion: Early high anion gap metabolic acidosis in patients with APAP overdose is self-limited and does not predict clinical or laboratory outcomes. Persistent or late metabolic acidosis in the absence of liver failure is not likely due to APAP and should prompt a search for other causes of metabolic acidosis. Finally, APAP overdose should be considered in patients presenting to the emergency department with altered mental status, as this is a treatable condition when detected early.
(C) 2010
Presented as a poster at the General Assembly of American College of Chest Physicians; Zein J, Kakoulas C, Toubia N, Kinasewitz G. Early high anion gap metabolic acidosis in patients with acetaminophen overdose: clinical significance. Chest 2004;126:872S.
* Corresponding author. Division of Pulmonary and Critical Care,
Department of Internal Medicine, Kings County Hospital Center, Brooklyn, NY 11203, USA.
E-mail address: [email protected] (D.J. Wallace).
Introduction
The liver is capable of substantial lactate utilization through both oxidation and gluconeogenesis. Impairment of these pathways is commonly listed as a cause of lactic acidosis. If left untreated, severe acetaminophen (APAP)
0735-6757/$ - see front matter (C) 2010 doi:10.1016/j.ajem.2009.04.005
toxicity results in fulminant hepatic failure with associated metabolic acidosis resulting from incompetent hepatic lactic acid utilization. Lactic acidosis in these patients is usually associated with hypoglycemia, which reflects a deficit in hepatic gluconeogenesis as well. In this setting, lactic acidosis is usually resistant to treatment and associated with a poor prognosis [1]. However, in contrast to the well-recognized scenario of APAP hepatic necrosis leading to lactic acidosis several days after ingestion, metabolic acidosis and altered mental status can also develop within hours of APAP overdose before any overt hepatotoxicity [2-4].
Co-ingestants are one explanation for the appearance of early metabolic acidosis; overdoses with tricyclic antide- pressants, barbiturates, salicylate, and nonsteroidal anti- inflammatory drugs [5] are known to be associated with a high anion gap metabolic acidosis. An independent association between APAP and early high anion gap metabolic acidosis (EHAGMA) has also been suggested [3], through both an impaired mitochondrial respiration mechanism [6,7] and depletion of liver glutathione stores leading to 5-oxoprolinuria production [8]. We hypothesized that APAP toxicity would be associated with metabolic acidosis in the absence of co-ingestants and that the severity of the acidosis would correlate with an increased APAP dose ingested, a higher plasma level, and a worse clinical outcome.
Methods
A retrospective chart review was conducted on a cohort of patients with APAP overdose, identified from hospital discharge International Classification of Diseases, 9th Edition, codes e850.4 and 965.4. These patients presented to either the University of Oklahoma Health Sciences Center or Saint Vincent’s Hospital and Medical Center of New York between January 1998 and November 2003. Only those patients with a primary discharge diagnosis of APAP overdose were included. Patients with ingestions occurring more than 24 hours prior were excluded, as were patients with a history of concomitant alcohol, tricyclic antidepressants, salicylate, barbiturate, or nonsteroidal anti- inflammatory drug ingestions. In addition, when available, patients with a detectible blood alcohol level or a urine drug screen positive for amphetamine, methamphetamine, barbiturate, benzodiazepine, cocaine, opiate, phencyclidine, or tricyclic antidepressant were excluded. Patients with a history of chronic liver disease, chronic renal insufficiency, and those with initial transaminase values higher than 3 times the upper normal values on admission were excluded. Patients were excluded if they had detectable plasma or urine ketones on admission; this decision was made to remove patients from the cohort who had simultaneous diabetic, starvation, or alcoholic ketoacidosis on arrival.
The anion gap was calculated as [Na+] - [Cl-] - [HCO-], with a normal value defined as 8 to 14 mEq/L. Patients were divided into 2 groups for comparison: high (>=15 mEq/L) vs normal anion gap at hospital presentation. Demographic (age, sex) and clinical data (hospital length of stay, the amount of APAP ingested, APAP serum level, mean arterial blood pressure, heart rate, temperature, arterial blood gas results, liver function tests, prothrombin time, the anion gap, creatinine, and lactic acid level) were collected for each patient, where available, from enrollment until hospital discharge. Altered mental status as documented by the initial treating physicians was also recorded. Hypotension and hypoglycemia were defined as mean arterial pressure less than 65 mm Hg and a plasma glucose level less than 70 mg/ dL, respectively. Patients were considered to have altered mental status if the words or derivatives of “lethargy,” “confusion,” or “coma” were included in the physician assessment from the emergency department (ED) or the initial evaluation of a consulting intensivist. Data abstraction was performed using a standardized data collection instru- ment by one of the study investigators.
Comparisons between groups were done using the 1-way
3
analysis of variance test (ANOVA), the Wilcoxon/Kruskal- Wallis, or Komogorov-Smirnov test when appropriate. Categorical variables were compared using the Pearson ?2 test. Data are presented as mean +- SDs or medians with 25% to 75% interquartile ranges (IQRs) where appropriate. A P value of less than .05 was considered statistically significant. Statistical analysis was performed using JMP statistical software (Version 4.0; SAS Institute Inc Cary, NC).
The study protocol was approved by the institutional review boards at both institutions.
Results
We identified 143 patients with APAP overdose; 69 with polysubstance overdose were excluded from analysis. Of the remaining 74 patients, 71 (96%) had a negative urine drug screen, and in 3 patients, this test was not ordered (Fig. 1). Baseline demographic and clinical information is
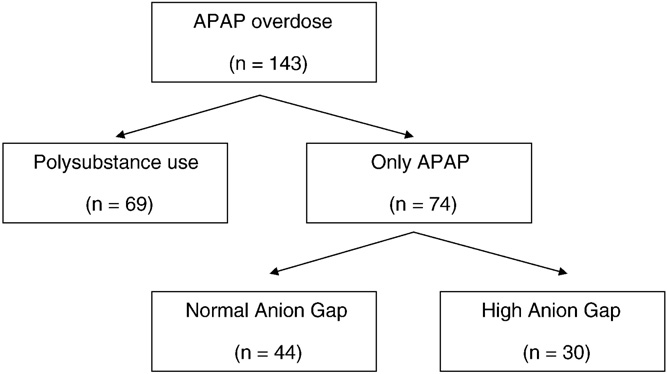
Fig. 1 Cohort derivation.
summarized in Table 1. Most patients took more than the potentially toxic dose of APAP (7.5 g in adults and 150 mg/ kg in children), with a median estimated ingestion of 13.0 g (range, 6.9-77 grams). Sixty-six patients (88%) had a toxic level according to the Rumach-Matthew nomogram, based on either a 4-hour level (n = 28) or a 12-hour level (n = 38); 9 patients (12%) had an uncertain timing of ingestion and were treated empirically. Most (47 patients, 64%) were admitted to the adult medical intensive care unit, whereas the remaining 27 patients were admitted to the children’s hospital. All patients presented within 24 hours of APAP intoxication and were treated with oral N-acetylcysteine at an initial dose of 140 mg/kg followed by 70 mg/kg every 6 hours for up to a total of 17 doses.
Sex, hospital length of stay, the amount of APAP ingested, and APAP serum level were not significantly different between those with or without EHAGMA. The median age for the cohort was 24 years; the range was 5 to 62 years. No patients required vasopressor support. No patients mani- fested hypoxemia or required mechanical ventilation.
Early high anion gap metabolic acidosis was present in 41% of patients on admission and persisted for 1.5 +-
0.5 days. For patients presenting within 4 hours (n = 28), the mean APAP level at 4 hours after ingestion was 210 +- 54 mg/ dL (range, 159-241 mg/dL). For patients presenting within 12 hours (n = 66), the mean APAP level at 12 hours after ingestion was 72 +- 35 mg/mL; all 54 patients who had a sample drawn at this time had a level of more than 20 per deciliter. Although lactate levels were only available for a small subset of the overall cohort (n = 9), lactate level was increased in proportion to the APAP concentration (r2 = 0.75, P b .05) and anion gap was associated with an elevated lactate level (r2 = 0.66, P b .05). A higher lactic acid level that persisted for 1 day was present in the group of patients with high anion gap 4.5 +- 5.4 vs 1.3 +- 2.0 mmol/L (P = .04). Plasma bicarbonate (HCO3) was significantly lower in the high anion gap group (20 mEq/L [IQR, 18-22] vs 24 mEq/L
Table 1 Baseline demographic and clinical data (means +- SD)
|
High (n = |
anion gap 30) |
Normal anion gap (n = 44) |
P |
|
|
HCO3 on day 1
|
20 |
(18-22) |
24 (22-24) |
.003 |
|
(mEq/L) |
||||
|
HCO3 on day 3 |
24 |
(22-26) |
24 (23-25) |
.62 |
|
(mEq/L) |
||||
|
Peak ALT (U/L) |
39 |
(30-72) |
33 (24-57) |
.12 |
|
Peak AST (U/L) |
25 |
(22-71) |
22 (18-42) |
.13 |
|
Peak total bilirubin |
0.65 (0.4-1.1) |
0.5 (0.3-0.8) |
.1 |
|
|
(mg/dL) |
||||
|
Peak PT (s) |
12.5 (11.8-13.8) |
12.7 (11.8-13.8) |
.8 |
|
|
Hospital length of |
3.5 (3-4.2) |
3.0 (2-4) |
.4 |
|
|
stay (d) |
||||

[IQR, 22-24], P = .003) but normalized within 2 days (24 mEq/L [IQR, 22-26] vs 24 mEq/L [IQR, 23-25], P =
Table 2 Laboratory and clinical data by anion gap grouping (medians and interquartile ranges)
.62). Plasma albumin level was normal in all patients (mean, 3.9 +- 1.7) and adjustment for anion gap was not required [9]. Early high anion gap metabolic acidosis was found in the absence of shock or liver failure.
In the overall cohort, 16 patients were described as “confused” and 13 were described as “lethargic.” Patients with increased anion gap had a higher incidence of confusion (48% vs 3%; P b .001) and lethargy (39% vs 6%; P = .003). None of the patients in the cohort were described as “comatose.”
The peak values of AST, ALT, total bilirubin, creatinine, and PT were similar in the normal and high anion gap groups (Table 2). On admission, the mean plasma glucose levels for the high anion gap and normal anion gap groups were 115 +- 26 and 110 +- 32 mg/dL, respectively (P = .52); no patient developed evidence of hypoglycemia during the hospitaliza- tion. There was no significant difference in the serum creatnine on either the day admission or in peak value for patients with high anion gap compared to those with a normal anion gap (Table 3).
 The first dose of N-acetylcysteine was administered within 8.9 +- 6 hours of ingestion (median, 7 hours; IQR, 5-12). None of the patients treated within 10 hours showed evidence of hepatic necrosis as manifested by a 3-fold increase in transaminases. Hepatic injury was evident in only one patient (1.5%) treated within 15 hours compared to 8 (88%) of 9 patients treated 16 to 24 hours after APAP ingestion. Three patients had a peak ALT level above 1000 U/L, and they all began treatment more than 16 hours after APAP ingestion.
The first dose of N-acetylcysteine was administered within 8.9 +- 6 hours of ingestion (median, 7 hours; IQR, 5-12). None of the patients treated within 10 hours showed evidence of hepatic necrosis as manifested by a 3-fold increase in transaminases. Hepatic injury was evident in only one patient (1.5%) treated within 15 hours compared to 8 (88%) of 9 patients treated 16 to 24 hours after APAP ingestion. Three patients had a peak ALT level above 1000 U/L, and they all began treatment more than 16 hours after APAP ingestion.
|
High (n = |
anion 30) |
gap |
Normal anion gap (n = 44) |
P |
||
|
Age (y) |
23 +- 2 |
29 +- 2 |
.045 |
|||
|
Sex (male %) |
37 |
23 |
.19 |
|||
|
Ingested dose (g) |
13 +- 1 |
10 +- 1 |
.16 |
|||
|
APAP level at 4 h |
204 +- |
18 |
188 |
+- 10 |
.4 |
|
|
(ug/mL) |
||||||
|
APAP level at 12 h |
79 +- 6 |
69 +- 6 |
.07 |
|||
|
(ug/mL) |
||||||
|
Mean arterial pressure |
86 +- 1 |
89 +- 1 |
.1 |
|||
|
(mm Hg) |
||||||
|
Heart rate (beats/min) |
91 +- 3 |
88 +- 3 |
.44 |
|||
|
Arterial oxygen |
97.7 +- 0.1 |
97.4 +- 0.2 |
.8 |
|||
|
saturation (%) |
||||||
|
Confusion (%) |
48 |
3 |
.001 |
|||
|
Lethargy (%) |
39 |
6 |
.003 |
|||
Table 3 Creatnine by anion gap grouping (means +- SD)
High anion gap Normal anion P
(n = 30) gap (n = 44)
Creatnine day 1 (mg/dL) 0.86 +- 0.28 0.93 +- 0.78 .62
Creatnine peak (mg/dL) 0.89 +- 0.27 0.97 +- 0.97 .64
There was no difference between patients treated within 10 hours or those treated 11 to 24 hours in terms of baseline anion gap (median, 13 [IQR, 11-15.5] vs 15 [IQR, 12-17];
P = .21), or baseline HCO3 (median, 22 [IQR, 20-24] vs 19
[IQR, 18-23]; P = .12), anion gap on day 3 (median, 12 [IQR, 10-14] vs 14 [IQR, 10-17]; P = .27), or HCO3 on day 3 (median, 24 [IQR, 23-25.5] vs 23.5 [IQR, 21-25.5]; P = .44).
The hospital length of stay was not different between the 2 groups (median, 3.5 [IQR, 3-4.2] vs 3 [IQR, 2-4]; P = .4).
Discussion
This is the largest study to examine the early acid-base disturbances that develop with APAP overdose. Previous investigators had observed the presence of an elevated anion gap early in the course of APAP poisoning, but case reports cannot establish disease incidence or prevalence [3,10]. One prospective series [11] noted that 12 (63%) of 19 patients seen early after APAP ingestion had an elevated anion gap on admission to the hospital, but many patients in this study had co-ingestants. We found that EHAGMA is relatively common in isolated APAP ingestions, occurring in 30 (41%) patients. Interestingly, the presence of EHAGMA was not predicted by either the 4-hour APAP level or the reported ingested dose.
In the subset of patients where lactic acid measurements were available, anion gap correlated with lactic acid levels. This finding was unlikely to reflect a type A lactic acidosis because no patients showed evidence of circulatory impairment or hypoxemia. Type B lactic acidosis secondary to hepatic dysfunction, which creates an imbalance between lactate production and utilization, is a more plausible explanation. In the overall cohort of patients, early high anion gap acidosis occurred in the presence of normal transaminases, bilirubin, and coagulation tests, suggesting that it was not related to direct hepatocellular necrosis. Although only 12 patients (16%) in this cohort had evidence of hepatic injury on arrival, our findings are consistent with previous reports that noted resolution of the lactic acidosis before the development of overt hepatic necrosis [11]. Further investigations are needed to elucidate the mechanism of EHAGMA in APAP overdose. Our investigation suggests that lactic acidosis may play a role, but this is not definitively established.
The EHAGMA observed in this cohort was self-limited in all patients, so any treatment beyond N-acetylcysteine administration and supportive care cannot be recommended. Patients who have persistent high anion gap acidosis and elevated lactate beyond the first 48 hours, however, require a detailed evaluation for other causes of metabolic acidosis. In the absence of liver failure, these conditions are not likely due to APAP.
In addition, many of the patients with mental status changes had an elevated anion gap (80%). For this reason,
APAP poisoning should be considered in the differential diagnosis of patients with metabolic acidosis of unknown etiology, in the setting of impaired mental status.
Limitations
There are several limitations to this study. (1) As is the case with all retrospective reviews, it is possible that selection bias may have compromised the internal validity of the results. Efforts were made to capture all patients discharged with a primary diagnosis of APAP overdose; however, some patients may have been missed. (2) As the chart reviewers were not blinded to the clinical chemistry results, unrecognized assessment bias may have influenced data abstraction. (3) All patients in this cohort were treated with oral n-acetyl cysteine (NAC). As such, the findings may not be generalizable to facilities using intravenous NAC. (4) Few conclusions can be made regarding the definitive role of lactic acidosis in the EHAGMA observed in this cohort because there were incomplete data for most patients. As such, this etiology of the EHAGMA remains speculative.
The importance of EHAGMA is incompletely under- stood. It is possible that this condition is clinically significant but that the described cohort study was not large enough to capture less frequent yet meaningful end points.
Conclusion
In this cohort of patients with APAP overdose, EHAGMA was self-limited and did not predict clinical or laboratory outcomes. Persistent or late metabolic acidosis in the absence of liver failure is likely not due to APAP and should prompt a search for other causes of metabolic acidosis. Finally, APAP overdose should be considered in patients presenting to the ED with altered mental status, as this is a treatable condition when detected early.
References
- Zabrodski RM, Schnurr LP. Anion gap acidosis with hypoglycemia in acetaminophen toxicity. Ann Emerg Med 1984;13:956-9.
- Zein J, Kakoulas C, Toubia N, Kinasewitz G. Early high anion gap metabolic acidosis in patients with acetaminophen overdose: clinical significance. Chest 2004;126:872S.
- Roth B, Woo O, Blanc P. Early metabolic acidosis and coma after acetaminophen ingestion. Ann Emerg Med 1999;33:452-6.
- Zezulka A, Wright N. Severe metabolic acidosis early in paracetamol poisoning. Br Med J (Clin Res Ed) 1982;285:851-2.
- Linden CH, Townsend PL. Metabolic acidosis after acute ibuprofen overdosage. J Pediatr 1987;111:922-5.
- Burcham PC, Harman AW. Acetaminophen toxicity results in site- specific mitochondrial damage in isolated mouse hepatocytes. J Biol Chem 1991;266:5049-54.
- Esterline RL, Ray SD, Ji S. Reversible and irreversible inhibition of hepatic mitochondrial respiration by acetaminophen and its toxic metabolite, N-acetyl-p-benzoquinoneimine (NAPQI). Biochem Phar- macol 1989;38:2387-90.
- Pitt JJ, Hauser S. Transient 5-oxoprolinuria and high anion gap metabolic acidosis: clinical and biochemical findings in eleven subjects. Clin Chem 1998;44:1497-503.
- Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med 1998;26:1807-10.
- Flanagan RJ, Mant TG. Coma and metabolic acidosis early in severe acute paracetamol poisoning. Hum Toxicol 1986;5:179-82.
- Gray TA, Buckley BM, Vale JA. Hyperlactataemia and meta- bolic acidosis following paracetamol overdose. Q J Med 1987;65: 811-21.